

FDA’s introduction to its rules for medical device regulation states: “Medical devices are classified into Class I, II, and III. Regulatory control increases from Class I to Class III. The device classification regulation defines the regulatory requirements for a general device type. Most Class I devices are exempt from Premarket Notification 510(k); most Class II devices require Premarket Notification 510(k); and most Class III devices require Premarket Approval.”1
Given that such a definition encompasses a large majority of medical products other than drugs, it is small wonder that medical device software now permeates a huge range of diagnostic and delivery systems. The reliability of the embedded software used in these devices and the risk associated with it has become a vital concern.
In response to that, the functional safety standard IEC 62304, “Medical device software – Software life cycle processes,” has emerged as an internationally recognized mechanism for the demonstration of compliance with the relevant local legal requirements. In practice, for all but the most trivial applications, compliance with IEC 62304 can only be demonstrated efficiently with a comprehensive suite of automated tools.2,3
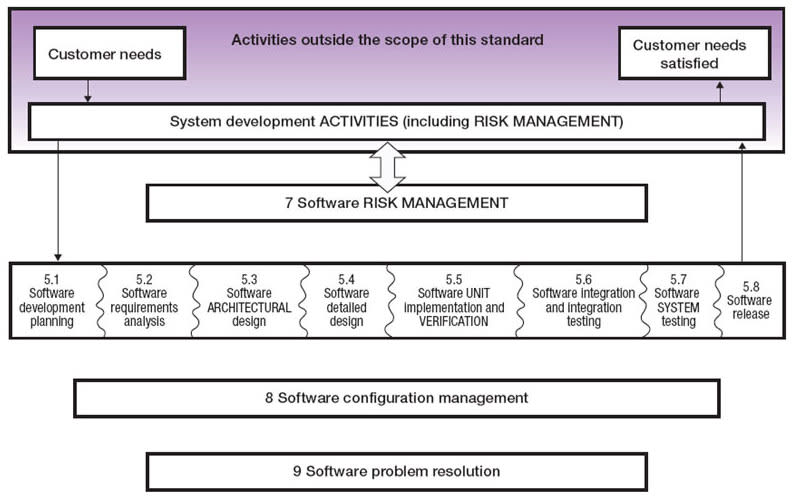
Part 1 of this article examines the development of detailed requirements and associated design of medical devices specified by IEC 62304, culminating in a detailed software design in accordance with clause 5.4 of the standard and shown in context in Figure 1.
Released in 2006, the IEC 62304 standard provides a framework of software development life cycle processes with activities and tasks necessary for the safe design and maintenance of medical device software. The processes, activities, and tasks described in clause 5 establish a common framework for medical device software life cycle processes that can be understood and shared within and between teams working on a project (see Figure 1).
IEC 62304 applies to the development and maintenance of medical device software when:
Software is itself a medical device.
Software is used as a component, part, or accessory of a medical device.
Software is used in the production of a medical device.
The IEC 62304 standard expects the manufacturer to assign a safety class to the software system as a whole, based on its potential to create a hazard that could result in an injury to the user, the patient, or other people.
There are three software safety classifications, as follows:
Class A: No injury or damage to health is possible.
Class B: Nonserious injury is possible.
Class C: Death or serious injury is possible.
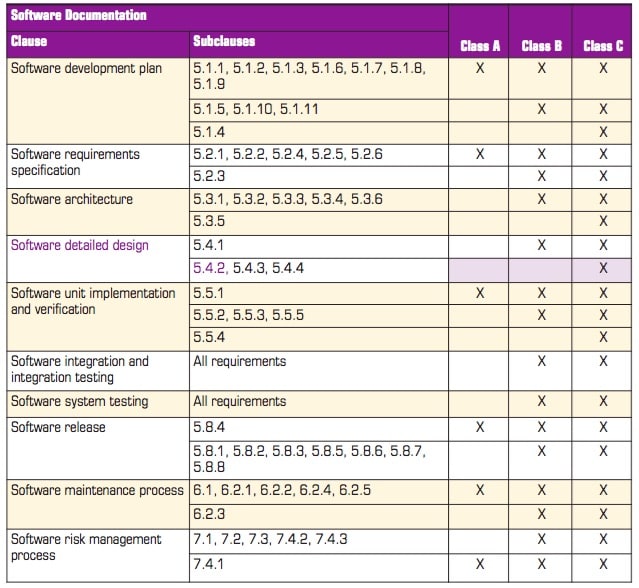
The classification assigned to a project has a tremendous impact on the code development process from planning, developing, testing, and verification through to release and beyond. It is therefore in the interest of medical device manufacturers to get it right the first time and avoid expensive and time-consuming rework.
In practice, any company developing medical device software will carry out verification, integration, and system testing on all software regardless of the safety classification, but the depth to which each of those activities is performed varies considerably. Table 1 is based on table A1 of the standard and gives an overview of what is involved.
For example, subclass 5.4.2 of the standard states that “The MANUFACTURER shall develop and document a detailed design for each SOFTWARE UNIT of the SOFTWARE ITEM.” Reference to the table shows that it applies only to Class C code and that it is not required for classes B and C.
IEC 62304 is essentially an amalgam of existing best practices in medical device software engineering, and the functional safety principles recommended by the more generic functional safety standard IEC 61508, which has been used as a basis for industry specific interpretations in a host of sectors as diverse as the rail industry, the process industries, and earth moving equipment manufacture.4
A process-wide, proven tool suite such as the LDRA tool suite, has been shown to help ensure compliance to such software safety standards (in addition to security standards) by automating both the analysis of the code from a software quality perspective as well as the required validation and verification work. Equally important, the tool suite enables life cycle transparency and traceability into and throughout the development and verification activities, facilitating audits from both internal and external entities. It is therefore a logical choice for working in accordance with IEC 62304.
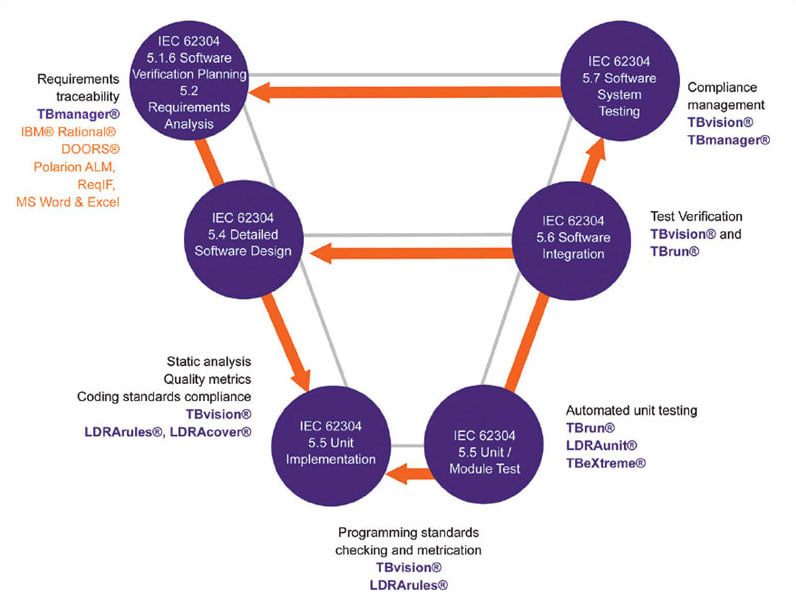
The V diagram in Figure 2 illustrates how the LDRA tool suite can help through the software development process described by IEC 62304. The tools also provide critical assistance through the software maintenance process (clause 6) and the risk management process (clause 7). Clause 5 of IEC 62304 details the software development process through eight stages ending in release. Notice that the elements of Clause 5 map to those in Figure 1 and Table 1.
5.1 Software Development Planning
Following the steps listed in Table 1 — and the V diagram in Figure 2 — the first objective is to plan the tasks needed for development of the software in order to reduce risks as well as to communicate procedures and goals to members of the development team. This helps ensure that system quality requirements for the medical device software are understood and met by all team members and can be verified within the project.
Development planning applies to all classes of devices. It involves the creation of the plan with reference to the system design and the definition of system requirements, and the selection and definition of the development standards, methods, and tools to be used. There should be a defined procedure for integration and integration testing. A format for documentation should be specified along with plans for configuration management and verification.
Thorough software development planning is vital to success. The foundations for an efficient development cycle can be established by using tools that can facilitate structured requirements definition, such that those requirements can be confirmed as met by means of automated document (or artifact) generation. IEC 62304 dictates that medical device software must include appropriate risk control measures in its system requirements.
The preparation of a mechanism to demonstrate that the requirements have been met will involve the development of detailed plans. A prominent example would be the software verification plan to include tasks to be performed during software verification and their assignment to specific resources.
5.2 Software Requirements Analysis
Once each system requirement has been identified in such a way as to make it possible to demonstrate traceability right from the system requirement through to software system testing, and to show this risk control measures have been accounted for, the next step is to derive and document the software requirements from the system requirements.
Achieving a format that lends itself to bi-directional traceability will help to achieve compliance with the standard. Bigger projects, perhaps with contributors in geographically diverse locations, are likely to benefit from an application life cycle management tool such as IBM® Rational® DOORS®, Siemens® Polarion® PLM®, or more generally, similar tools offering support for standard Requirements Interchange Formats. Smaller projects can cope admirably with carefully worded Microsoft® Word® or Microsoft Excel® documents, written to facilitate links up and down the V model.5–7
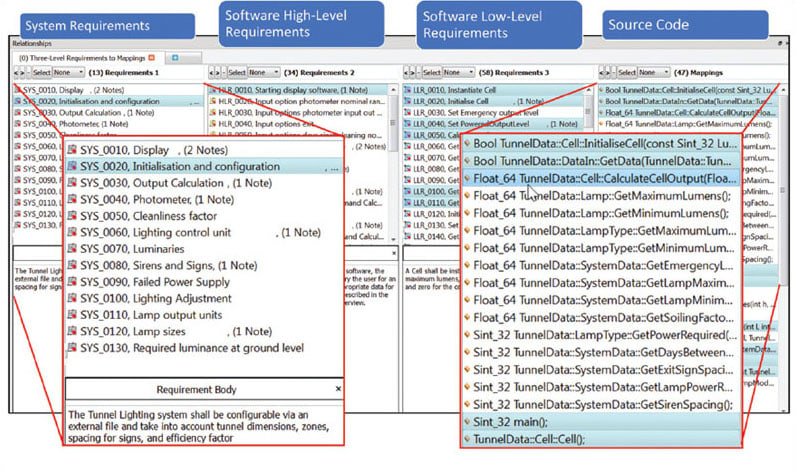
Requirements rarely remain unchanged throughout the lifetime of a project, and that can turn the maintenance of a traceability matrix into an administrative nightmare. A requirements traceability tool alleviates this concern by automatically maintaining the connections between the requirements, development, and testing artifacts and activities. Any changes in the associated documents or software code are automatically highlighted such that any tests required to be revisited can be dealt with accordingly (see Figure 3).
5.3 Software Architectural Design
The software architectural design activity requires the manufacturer to define the major structural components of the software, their externally visible properties, and the relationships between them. Any software component behavior that can affect other components should be described in the software architecture, such that all software requirements can be implemented by the specified software items. This is generally verified by technical evaluation.
Since the software architecture is based on the requirements, developing the architecture means defining the interfaces between the software items that will implement the requirements. Many of these software elements will be created by the project, but some may be brought in from other projects even in the form of third-party libraries. Such code must be shown to comply with the project’s functional and performance requirements.
In addition, some architectural elements may need to be segregated for risk management purposes, such that their connections to the other elements then become part of the overall architecture. Tools can help in many elements of the architectural design process, including requirements specification, design model traceability, and verification.
If a model-based approach is taken to software architectural design — for example, using MathWorks® Simulink®, IBM Rational Rhapsody®, or ANSYS® SCADE — then a tool suite that is integrated with the chosen modelling tools will make the analysis of generate code and traceability to the models far more seamless.8–10
5.4 Software Detailed Design
Once requirements and architecture are defined and verified, detailed design can be built on this foundation. Detailed design specifies algorithms, data representations, and interfaces between different software units and data structures. Because implementation depends on detailed design, it is necessary to verify the detailed design before the activity is complete, generally by means of a technical evaluation of the detailed design as a whole, and verification of each software unit and its interfaces.
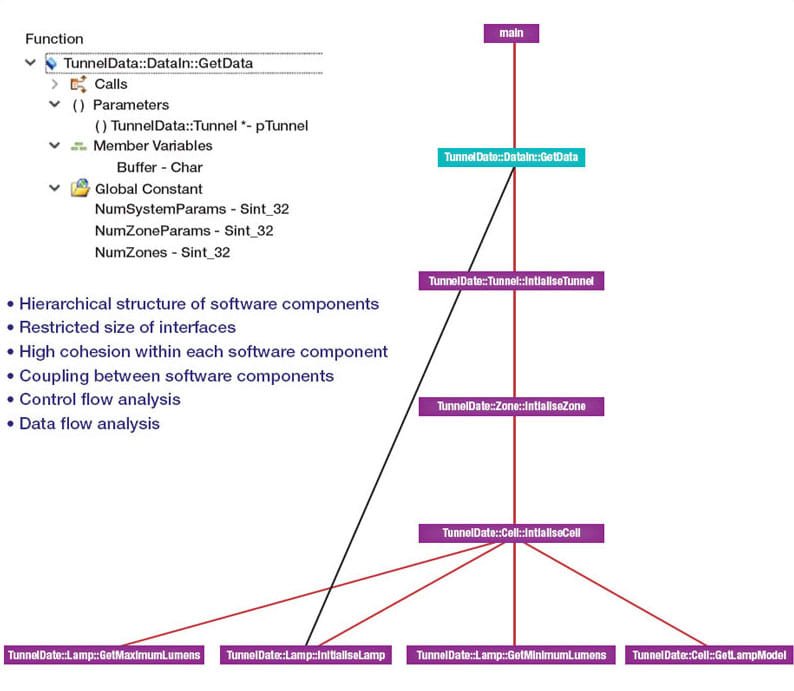
Later in the development cycle, a tool suite can help by generating graphical artifacts suited to the review of the implemented design by means of walkthroughs or inspections. One approach is to prototype the software architecture in an appropriate programming language, which can also help to find any anomalies in the design. Graphical artifacts like call graphs and flow graphs, are well suited for use in the review of the implemented design by visual inspection (see Figure 4).
Part 2 of this article will look at the second part of the project life cycle involving the implementation of the now-established design in the form of source code. It will consider how static analysis can help to ensure that coding rules are adhered to, helping to minimize errors in the developed application. It will discuss how dynamic analysis in the form of system and unit tests show that the design has been implemented correctly and completely, and that the software has been properly exercised during test and prior to release. Finally, it will outline how the influence of IEC 62304 extends to the maintenance and modification of the product once out in the field.
This article was written by Mark Pitchford, Technical Specialist for LDRA (Wirral, UK). For more information, Click Here .

