
Medical devices manufactured today require significant attention to safety and human factors engineering that has not always been exercised in medical device design. IEC safety standards mandated by the general standard, IEC 60601-1, updated to the 3rd edition, and now Amendment No. 1 to that standard, require testing and verification of devices and essential performance, while particular standards focused on specific types of devices add considerations that must be met.
Background
The advent of medical device regulation brought on the need for standardizing the ways a manufacturer could demonstrate the safety and effectiveness of medical devices. Voluntary safety standards developed through the input and efforts of industry stakeholders helped to address some of this need. These safety standards, wholly or partially recognized by regulatory authorities around the world, generally prescribe requirements for design, testing, and performance. In practice, upon request from manufacturers, independent third party certification bodies follow these standards by performing design reviews and programs of tests to evaluate the extent of compliance with requirements. That said, manufacturers with the requisite expertise could perform their own evaluations and tests as part of internal procedures.
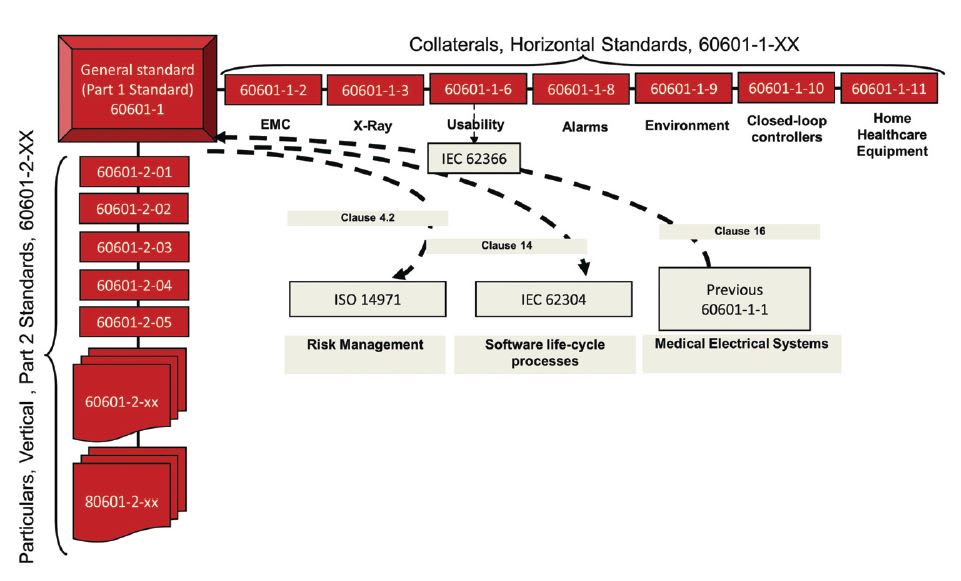
As medical device technology evolved, so too did the scopes and requirements of standards. For example, standard UL 544 (Standard for Medical and Dental Equipment), issued in 1972, set forth requirements for electrical medical and dental equipment intended for professional use by personnel in hospitals, nursing homes, medical care centers, medical and dental offices, and similar facilities. UL 544 gave way to the internationally harmonized standard IEC 60601- 1 (Medical Electrical Equipment, Part 1: General Requirements for Safety), issued in 1988, which broadened the overall scope of requirements and arrived with a large family of supplemental standards known as collateral standards (IEC 60601-1-X) and particular standards (IEC 60601-2-XX). (See Figure 1)
Collateral standards, also known as horizontal standards, cover general cross-cutting aspects such as electromagnetic compatibility (IEC 60601-1-2) or usability (IEC 60601-1-6) or alarms (IEC 60601-1-8). Particular, or vertical, standards apply to devices with specific intended uses and technologies, like endoscopic equipment (IEC 60601-2-18), electrocardiographic monitoring equipment (IEC 60601-2-27), or surgical luminaires (IEC 60601-2-41).
Subsequently, the 3rd and current edition of IEC 60601-1 (Medical Electrical Equipment, Part 1: General Requirements for Basic Safety and Essential Performance) followed in 2005. The noteworthy change in the title for the 3rd edition added the term “essential performance,” which the standard originally defined as “performance necessary to achieve freedom from unacceptable risk”. This signaled a clear shift to incorporate risk management, prescribed by ISO 14971 principles, as a vital requirement in safety standards. Also, in contrast to the preceding standards, the 3rd Edition contemplates device use not only by trained healthcare professionals, but also by nonprofessionals, such as the patients themselves or their caregivers. The 3rd edition relies on collateral standard IEC 60601-1-11 to address these latter users of home healthcare equipment.
While the demand for improving the versatility of healthcare fosters innovation, it also results in challenges to ensuring patient and user safety. New technology can raise new questions of safety and effectiveness, yet consensus thinking may not be readily clear among regulators and industry stakeholders. Nevertheless, while underscoring the need for risk-based standards and design principles, the ever-evolving complexity of medical devices has led to a relatively recent and increasing emphasis on human factors engineering.
Human Factors Engineering (HFE)

FDA regulations and IEC standards place an expectation on manufacturers to apply human factors engineering to the product development process to manage use-related risks with the product. HFE activities performed during product development will assess the potential risks with the product and evaluate whether or not the associated design mitigations effectively reduce or eliminate the risk.
Including HFE into the entire design and development process involves, but is not limited to, performing the following activities:
- Defining the device’s intended users
- Defining the environments in which the product is used
- Defining the primary and frequent functions users will perform with the system
- Defining the tasks that users will perform with the device
- Identifying users’ needs
- Developing a list of user interface design requirements to meet users’ needs
- Defining the primary and frequent functions users will perform with the system
- Identifying use-related risks and their associated likelihoods and severities
- Identifying risk control measures (i.e., mitigations to the risks), which might include design features and learning aids.
- Conducting formative evaluations to assess the device’s safety and usability
- Conducting a validation test to provide evidence that the device is safe and effective for users
Ultimately, manufacturers must validate that the product’s users can employ the device safely and effectively in the intended use environments.
Conclusion
Medical technology is rapidly advancing. Manufacturers are faced with the challenge of meeting the ever-increasing needs of the medical community to keep pace with exciting outcomes from medical research and treatment advances. However, it is necessary that products maintain a high degree of usability while minimizing use-related risk.
During device design, by paying close attention to the factors that are identified through safety testing, and usability concerns that can arise during the HFE activities identified above, human factors specialists are able to meet the expanding expectations of medical device users. Risk management activities are interwoven throughout the design process and underpin successful safety and HFE certification.
Manufacturers might find that they can streamline the overall design process for a given medical device by partnering early with safety testing houses and HFE service providers. Early engagement helps identify and manage potential safety, usability and/or essential performance issues, allowing timely resolution within the product development lifecycle.
This article was written by Linda Chatwin, North American Business Manager, Medical Regulatory Advisory Services, UL, LLC, Camas, WA; Maureen Mulcare, Managing Human Factors Specialist II, UL, LLC, Concord, MA; and Jeff Rongero, Staff Engineer, Health Sciences, UL LLC, Research Triangle Park, NC. For more information, Click Here .

